Epigenetic Regulation in Plants
Group leader Dr Maike Stam
Epigenetic and chromatin regulation of gene expression
The tremendously rapid advances in high-throughput sequencing, but also advances in other techniques have enabled many exciting new discoveries in the field of gene regulation over the past years. We use such techniques to study gene regulation in plants. Our model systems are maize, tomato, Arabidopsis Thaliana, and oilseed rape. Two research lines are our main focus:
- Gene regulation by epigenetic mechanisms and chromatin structure
- Control of gene expression by cis-regulatory sequences
Regulation by epigenetic mechanisms & chromatin structure
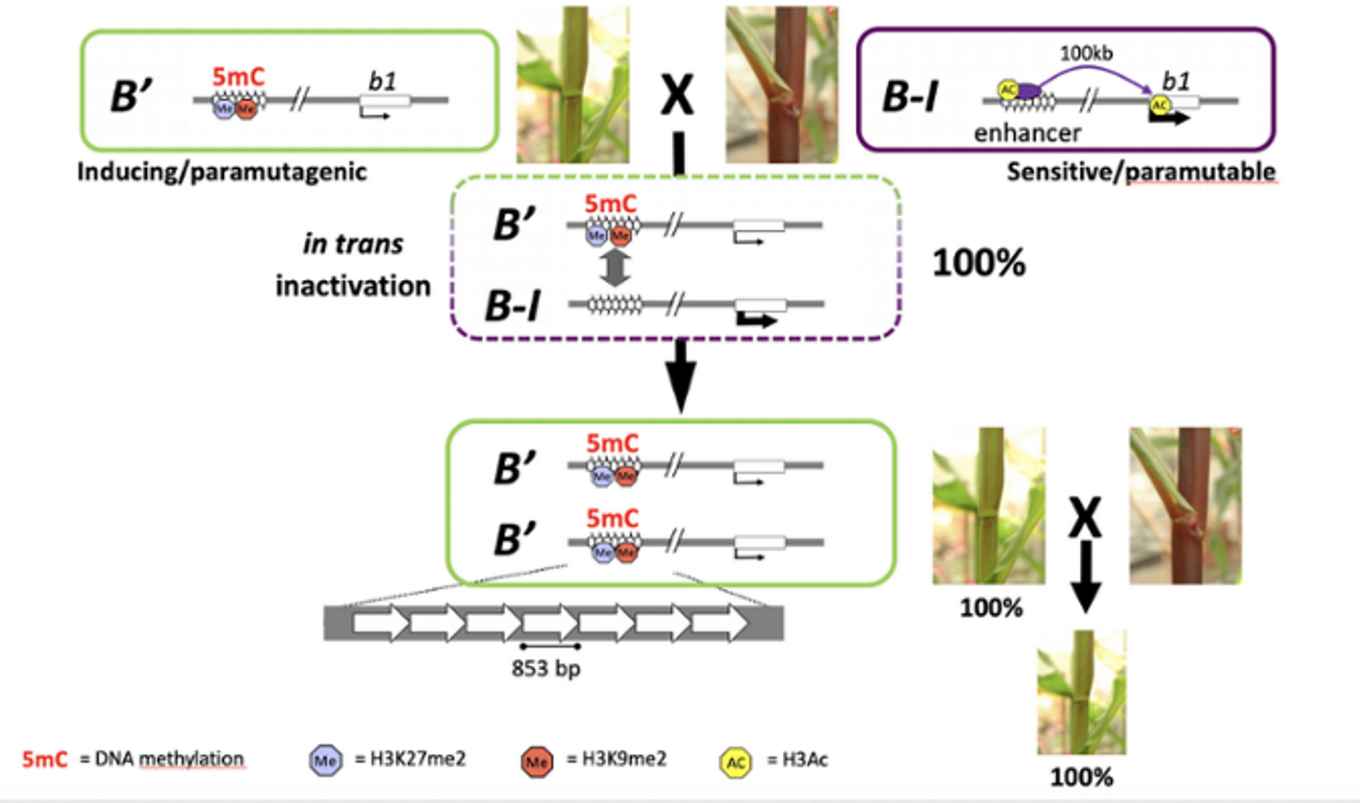
Plant and animal breeding has been done for thousands of years. It forms the basis of food production and many other human activities. Breeding is based on the heritability of desirable traits and the reiterated process of crossing and selection. Most differences in heritable traits are due to genetic variation, which are DNA sequence differences between individuals. But importantly, in addition to that well-known option, epigenetic variation can result in trait variation and contribute to the diversity we know of today. Epigenetic variation refers to mitotically and/or meiotically heritable changes in genome function based on chromatin modifications instead of changes in the DNA sequence. Examples of such modifications are DNA methylation and histone modifications. Epigenetic modifications are heritable, but not as stable as the genetic code. At particular DNA sequences the epigenetic information can be remodeled, among others under the influence of homologous DNA sequences carrying different epigenetic marks. The latter is well known to occur between individual alleles in a process called paramutation, but has also been indicated to occur at a genome-wide scale in heterosis, a phenomenon extensively exploited in agricultural breeding in which F1 hybrids between two parents display superior phenotypes compared to that of their parents. Currently, the contribution of epigenetic variation to beneficial traits displayed by crop plants and livestock, as well as the molecular basis underlying heterosis are still largely unknown. The aim of this research line is to unravel the mechanisms underlying the transfer of epigenetic information, both between specific alleles as genome-wide.
Control of gene expression by cis-regulatory sequences
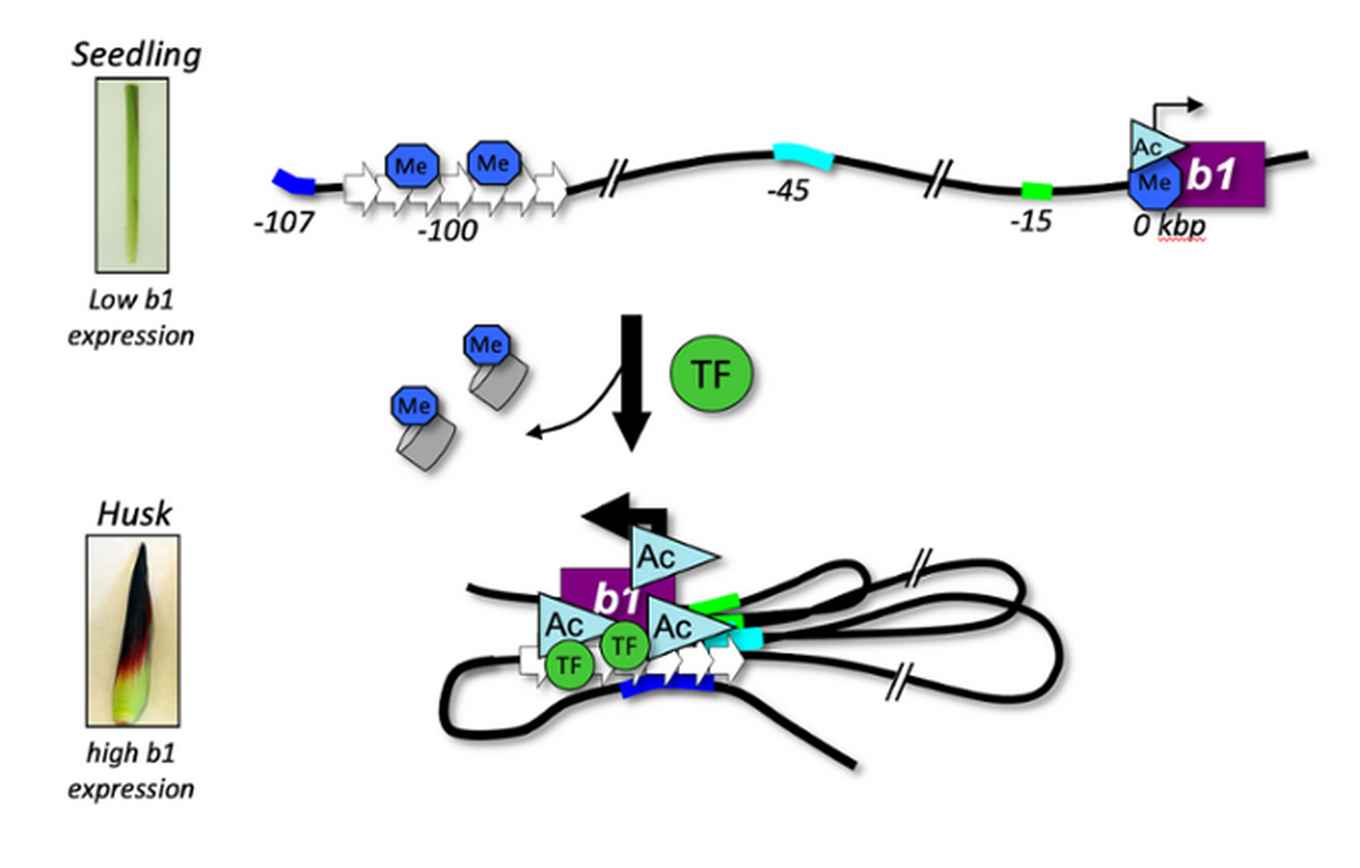
The differentiation of cells and their response to external signals such as light, temperature and pathogens is largely accomplished through the activation and repression of regulatory DNA sequences such as transcriptional enhancers at the correct moment in time and space (Schmitz et al, 2022). Enhancers are non-coding DNA sequences that activate the transcription of genes located up to several Mb away. Knowledge on the transcriptional regulatory code is crucial for the functional annotation of the genome and the selection of desirable traits for plant breeding. The regulatory code of plants, especially that of crop plants, is however still largely unknown. Genome-wide approaches have identified thousands of enhancers in animals, but are only very recently being applied to plants. This line of research is focused on unravelling the regulatory code of crop plants in general, and that of Zea mays in particular. Current and future work is focused on the identification and characterization of regulatory sequences and their target genes. This work contributes to a better understanding of the regulatory code in plants, and will enlarge the toolbox for the functional annotation and characterization of complex genomes, and promises new targets for informed breeding in crop plants.